py-sc-fermi¶
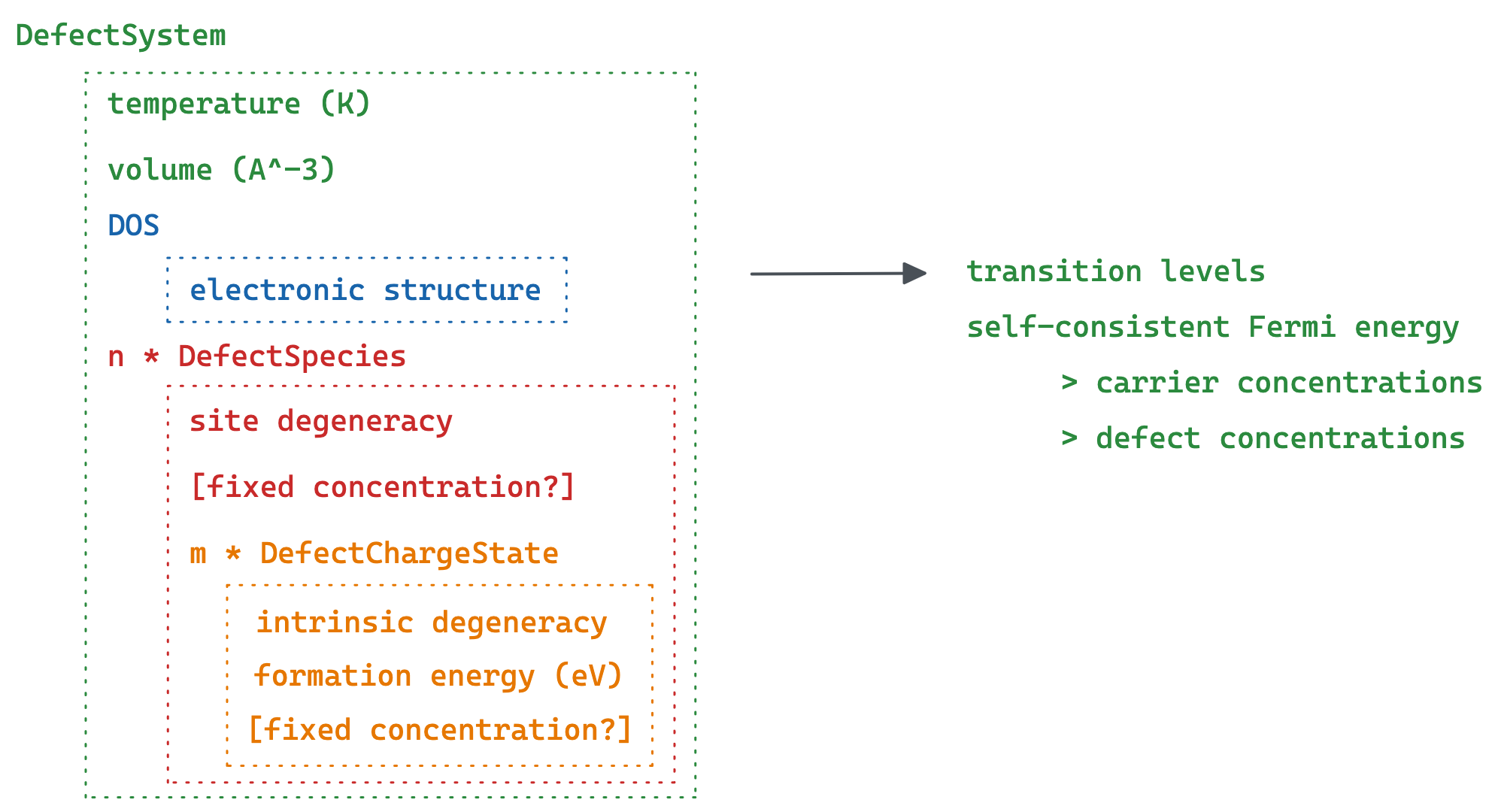
py-sc-fermi is an open-source Python package for calculating the concentration of point defects in (semiconducting) crystalline materials.
The required inputs are the volume, density of states of the bulk material, and the formation energies and degeneracies of the point defects.
The outputs include the self consistent Fermi energy, defect transition levels, and concentrations of the point defects, electrons and holes at a given temperature.
py-sc-fermi uses a numerical method to solve for the self-consistent Fermi level in a material, necessary for accurately quantifing the populations of point defects in such materials.
The approach used in this code was initially based off the algorithm used by the FORTRAN code SC-Fermi, as described in this Paper.
Papers that use py-sc-fermi¶
Haouari et al., Impact of Solution Chemistry on Growth and Structural Features of Mo-Substituted Spinel Iron Oxides, 2021, 10.1021/acs.inorgchem.1c00278
Squires et al., Low Electronic Conductivity of Li7La3Zr2O12 Solid Electrolytes from First Principles, 2022, 10.1103/PhysRevMaterials.6.085401
Jackson, Parret and Willis et al., Computational Prediction and Experimental Realization of Earth-Abundant Transparent Conducting Oxide Ga-Doped ZnSb2O6, 2022, 10.1021/acsenergylett.2c01961
Cen et al., Cation disorder dominates the defect chemistry of high-voltage LiMn1.5Ni 0.5O 4(LMNO) spinel cathodes, 2023, 10.1039/D3TA00532A
Nicolson et al., Cu2SiSe3 as a promising solar absorber: harnessing cation dissimilarity to avoid killer antisites, 2023, 10.1039/D3TA02429F